The Evolving Landscape of EU Centralised Marketing Authorisations
Published May 12, 2026
Published 29th April 2024
The FDA published a draft guideline on the potency of Cellular and Gene Therapy (CGT) in December 2023. The draft guidance describes the FDA’s recommendations for developing a potency assurance strategy through manufacturing process design, material controls, in-process control, and release testing to ensure the product’s potency. This blog covers the key points to meet these recommendations and potential challenges.
Potency is the quantitative measure of biological activity and is one of the critical quality attributes (CQAs) for medicinal products of biological origins. While potency testing remains essential, the new FDA draft guideline aligns with the existing principles of Quality Risk Management (ICH Q9 (R1)) and recommends developing a comprehensive potency assurance strategy for CGT products. The key points and, consequently, the impact on regulatory filings are highlighted in this article.
The deadline for comments is now closed, and the FDA is working on the document’s final version, which will eventually replace the 2011 guidance on Potency Tests for Cellular and Gene Therapy Products when finalised.
The FDA recommends using the approach already described in the existing ICH Pharmaceutical Development Guideline (ICH Q8 (R2)) for potency. To develop a potency assurance strategy, it is important first to understand the product’s potency-related characteristics and then perform a Quality Risk Management (ICH Q9 (R1)) analysis to assess and minimise the risks to potency.
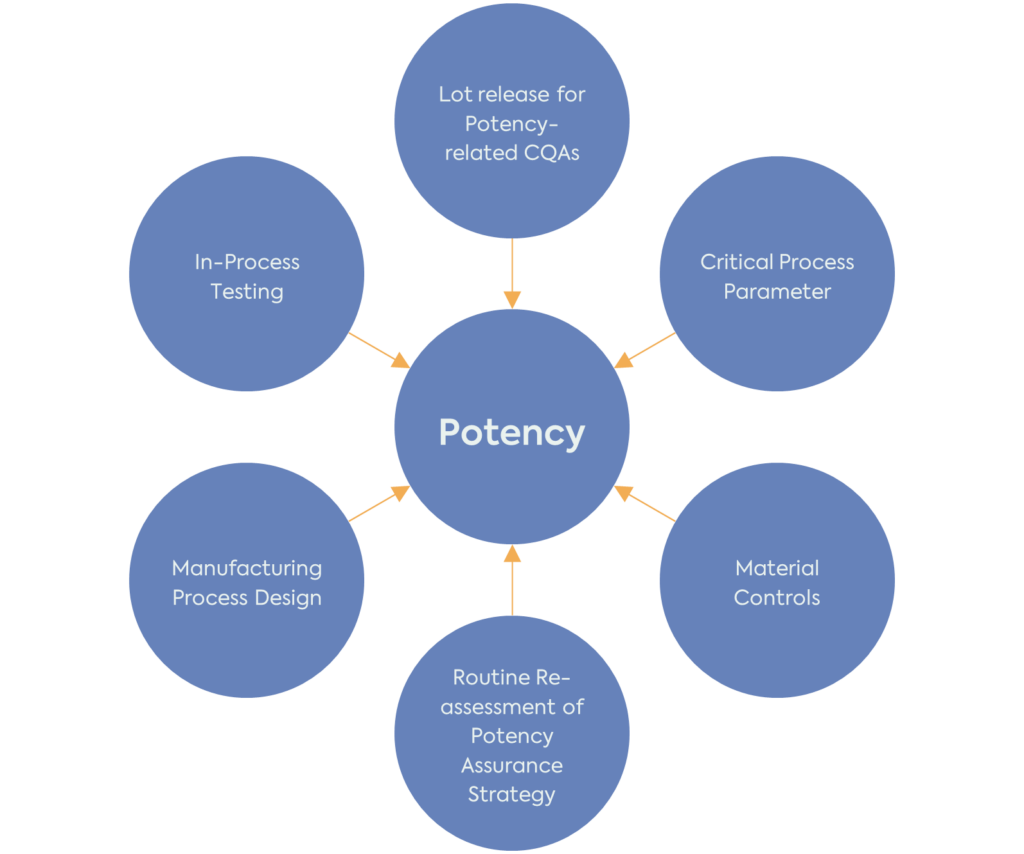
It is important to note that the potency assurance strategy should be reassessed and refined throughout the product development. Potency-related CQAs, risk assessment and strategy for reducing the risk to these CQAs should be in place before the start of clinical investigations. However, the product’s QTPP, CQAs, CPPs and potency assurance strategy will evolve with the increased understanding of the product and manufacturing process.
With the implementation of the guidance, the following additional information will be required in Module 2 and Module 3 of an Investigational New Drug (IND) and Biologics License Application (BLA) filing.

The second part of the guideline focuses on the potency assay itself. Although the potency assurance strategy provides other tools for controlling the potency of the product, potency assays are still an essential part of the strategy. Assays measuring potency-related CQAs are used for the release of the product, during stability studies to establish the product shelf-life or during in-use stability studies to ensure the product remains potent during administration. Potency assays are also essential to demonstrate comparability of the product lots to support manufacturing changes and during manufacturing process development to identify the steps that can affect the product potency.
Potency testing is a complex yet common challenge for developers of CGT products. Moreover, the FDA often raises challenging queries related to the potency when an IND application is submitted. This draft guideline provides valuable recommendations on potency tests and for developing a complementary, broader approach to ensure potency of the product.
DLRC’s team of CMC and biologic experts can advise on current regulations on potency testing and author and/or review CMC filings. Please get in touch with us at hello@dlrcgroup.com for further information.


Published May 12, 2026

Published May 07, 2026

Published Mar 20, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025