The Evolving Landscape of EU Centralised Marketing Authorisations
Published May 12, 2026
Published 23rd July 2024
Annex XVI of the European Union Medical Device Regulations (MDR) has introduced many new requirements for devices with no medical purpose in the EU. This article provides manufacturers of Annex XVI devices with an overview of the regulations, details the product requirements within and offers practical tips for their implementation. This article is beneficial not only to manufacturers of Annex XVI devices but to all medical device specialists to understand this new area of the regulations in the EU and how it can be navigated.
In 2017 the MDR was introduced into the EU. This new legislation repealed and replaced the Medical Device Directive 94/42/EEC (MDD) and the Active Implantable Medical Device Directive 90/385/EEC (AIMDD), causing a significant shift in the EU’s approach to medical device regulation. The EU undertook the overhaul of these directives because they did not cover many of the technology advances that have occurred in the last two decades. The introduction of the MDR has updated the 30 year old legislation surrounding medical devices with new requirements including enhanced clinical data, strengthened post market surveillance data, quality management system requirements and more. The European Parliament passed the MDR and began the transition period in May 2017 with enforcement beginning in May 2021.
Annex XVI devices are specific product types that do not have an intended medical purpose. There were previously no specific requirements for the Annex XVI products in the EU and, crucially for Annex XVI device manufacturers. These products cannot be ‘grandfathered in’, which means that manufacturers cannot use existing certification to justify compliance to the new requirements. All new requirements found in the MDR will be applicable to products within the scope of the regulation, including those already on the market.
An Annex XVI product is a category of products for which a manufacturer claims only an aesthetic or non-medical purpose. Annex XVI products have been introduced into the MDR due to the similarity of these products to medical devices. These products do not have a physiological, biological or metabolic action on the body, their action is mechanical in nature like medical devices. Annex XVI products’ functionality and risk profiles are also comparable to medical devices.
Contact lenses or other items intended to be introduced into or onto the eye. This does not include solutions for disinfecting, cleaning and rinsing contact lenses.
Devices intended to be totally or partially introduced into the human body through surgically invasive means for the purpose of modifying the anatomy or fixation of body parts with the exception of tattooing devices and piercings. This does also not include products for keratopigmentation.
Substances, combinations of substances, or items intended to be used for facial or other dermal or mucous membrane filling by subcutaneous, submucous or intradermal injection or other introduction, excluding those for tattooing. This category does not include equipment for transferring hair follicles in the context of hair transplant surgery (follicular injection device).
Equipment intended to be used to reduce, remove or destroy adipose tissue, such as equipment for liposuction, lipolysis or lipoplasty. This does not include a product intended for anything other than adipose tissue.
High intensity electromagnetic radiation (e.g. infra-red, visible light and ultra-violet) emitting equipment intended for use on the human body, including coherent and non-coherent sources, monochromatic and broad spectrum, such as lasers and intense pulsed light equipment, for skin resurfacing, tattoo or hair removal, or other skin treatment. This does not include products such as serums, creams or gels to be used before and/or after the hair removal treatment.
Equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain. This group only includes products which apply an electric current to the cranium.
The introduction of these product groups into the MDR is welcome news for healthcare professionals where such products have been known to cause serious clinical issues for users. Common issues with these product types are presented in Table 1 below.
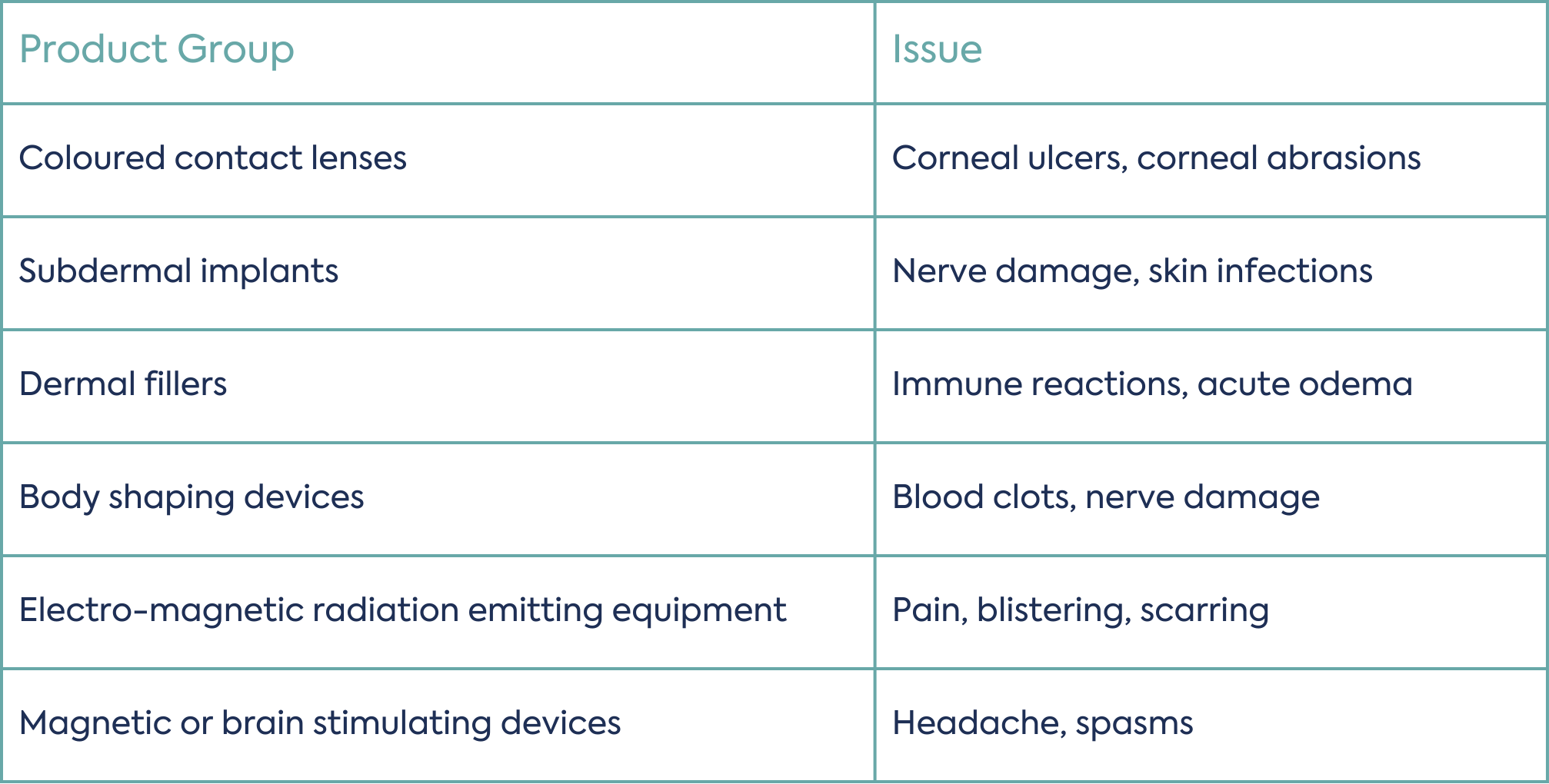
Inclusion of these previously unregulated groups of products into the scope of the MDR introduces new requirements around manufacturing and surveillance to reduce such clinical issues and ultimately protect the health and safety of users.
When considering market entry for an Annex XVI product in the EU, many of the elements of the traditional regulatory pathway for a ‘medical device’ are applicable to Annex XVI products. Understanding the requirements of the MDR is therefore critical for Annex XVI product manufacturers. A key requirement, found in Recital 12 of the MDR, states that a medical device without an intended medical purpose is to be governed by Common Specifications. Once a Common Specification becomes applicable the additional contents of the MDR are also applicable to Annex XVI devices.
In December 2022, four and a half years after the MDR was published, the EU released two Commission Implementing Regulations. These implementing regulations cover Common Specifications for all Annex XVI devices and classification requirements around active Annex XVI products.
In addition to Common Specifications, the MDR introduces clinical data collection and the requirement for a clinical evaluation plan and report, as well as the considerations for risk management for the device. There is the introduction of Quality Management System (QMS) requirements encompassing manufacture and management of the devices as well as post market surveillance activities.
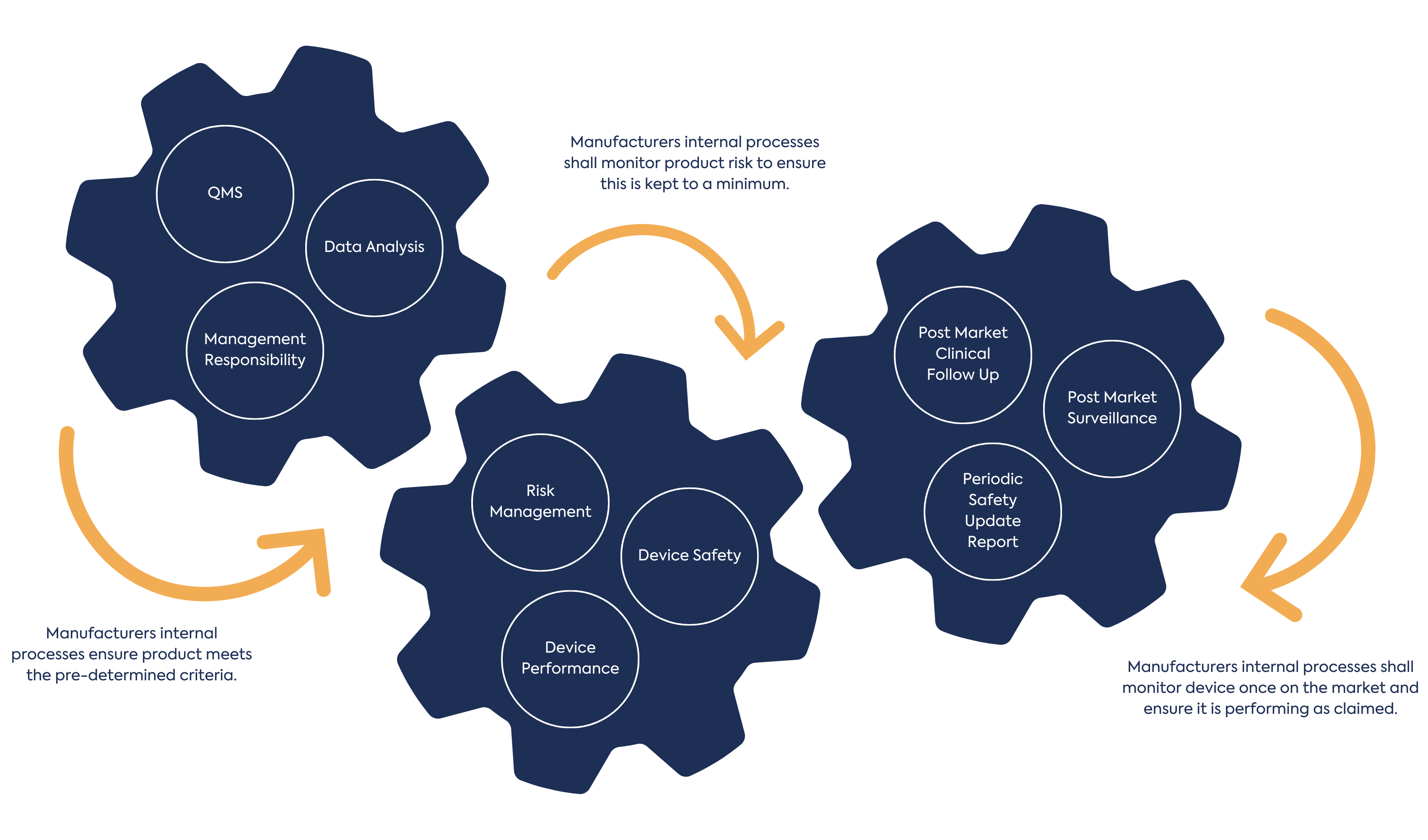
The application of this regulation to Annex XVI devices will be nuanced, as the implementation of the regulation will be through the lens of the Annex XVI criteria i.e. having no medical purpose. Understanding all applicable elements of the MDR and how they relate to Annex XVI products is critical to device development.
The following sections of this article elaborate on key elements of the MDR and how they should be considered when dealing with an Annex XVI device.
Annex I of the Implementing Regulation (EU) 2022/2346 includes general risk management considerations as well as specific product safety information for all Annex XVI products. Annex II to Annex VII of the Implementing Regulation details the product specific considerations which must be made for each product group. The requirements include labelling, risk, design and manufacturing elements. Manufacturers seeking regulatory compliance or certification for Annex XVI products should apply all elements within the Common Specification (unless an adequate justification can be provided) to ensure regulatory compliance.
For MDR Annex XVI devices the classification is governed by the rules set out in MDR Annex VIII, however, not all rules can be applied. Rules 9 and 10 (which are the rules for active therapeutic and diagnostic devices) assume a medical purpose within the text of the rule and can therefore not be used. Therefore, Commission Implementing Regulation (2022/2347) was adopted for the reclassification of certain Annex XVI products:
Classification of the device will determine its conformity assessment route:
For all Annex XVI devices, manufacturer’s design controls must meet the requirements within the MDR (MDR Article 10 (9, g)), however this will also include the applicable standard (ISO 13485 clause 7.3) if the device is Class I (sterile, measuring or reusable), Class IIa, Class IIb or Class III. Manufacturers are recommended to apply an ISO 13485 quality management system to their organisation as applicable. This will ensure suitable certification is granted for market entry.
The technical documentation to be compiled for an Annex XVI device should meet the requirements specified in Annex II of the MDR, including device description, benefit risk analysis, product verification and validation data and more. Manufacturers without experience of these types of files would benefit from utilising the guidance and templates released by the Medical Device Coordination Group to interpret and implement these additional requirements.
The approach to risk management by Annex XVI device manufacturers should meet the requirements of the MDR as well as those in the applicable standard EN ISO 14971: 2019. Also, manufacturers must consider product specific risks, found in the common specifications, as part of the risk management study.
In addition, for Annex XVI devices, the EU MDR states that risk acceptability shall be understood to mean that the device…‘when used under the conditions and for the purposes intended, should not present a risk at all or presents a risk that is no more than the maximum acceptable risk related to the product’s use which is consistent with a high level of protection for the safety and health of persons’.
This criterion is different from that of traditional risk management ensuring that risks are removed completely or only as high as can be expected for the product type. Therefore, the risk profile for Annex XVI devices must be as close to zero as possible for compliance. Manufacturers of these products must ensure that the team completing this task provide expert product knowledge as well as QMS, regulatory and risk management experience.
Annex XVI device manufacturers must ensure that claims for these devices do not refer to a clinical benefit. The label must bear the words “non-medical purpose” followed by a description of that non-medical purpose. In addition, information for safety must highlight the different degree of understanding for consumers, when compared with healthcare professionals. Annex I of the MDR and the relevant product Annex (Annex II to Annex VII) within the Common Specification contain more information on labelling.
For clinical evaluation of Annex XVI devices, the requirement to ‘demonstrate a clinical benefit’ shall be understood to mean ‘demonstrate the safety and performance of the device’. The MDR provides further guidance with regards to Annex XVI devices:
‘Clinical evaluations of those products shall be based on relevant data concerning safety, including data from post-market surveillance, Post Market Clinical Follow Up (PMCF), and, where applicable, specific clinical investigation. Clinical investigations shall be performed for those products unless reliance on existing clinical data from an analogous medical device is duly justified.’
There is the option to utilise an equivalent product for demonstration of safety and performance. When considering the equivalence route to market for devices without an intended medical purpose, devices with a medical purpose cannot be considered equivalent. However, dual purpose devices with a medical and non-medical intended use can be used. Clinical, technical, and biological attributes must be compared to the non-medical intention only. For details on how the clinical characteristics are compared for Annex XVI devices please see Table 2 below:

The need to complete a clinical investigation for the device will depend on the nature of the device and its intended use. To determine if a clinical investigation is necessary the clinical strategy for the device must be considered as early as possible in the development process.
Annex XVI device manufacturers must use the guidance found in Annex III of the MDR[1] for PMS of Annex XVI products. Manufacturers are recommended to review existing processes for compliance against appropriate guidance and the MDR requirements.
The sum total of these new requirements for Annex XVI products equates to a considerable amount of work for manufacturers in order to ensure new or continued regulatory compliance. In addition to this workload, there is a complex set of timelines for the transition of existing products to the MDR. The timelines are summarised below:
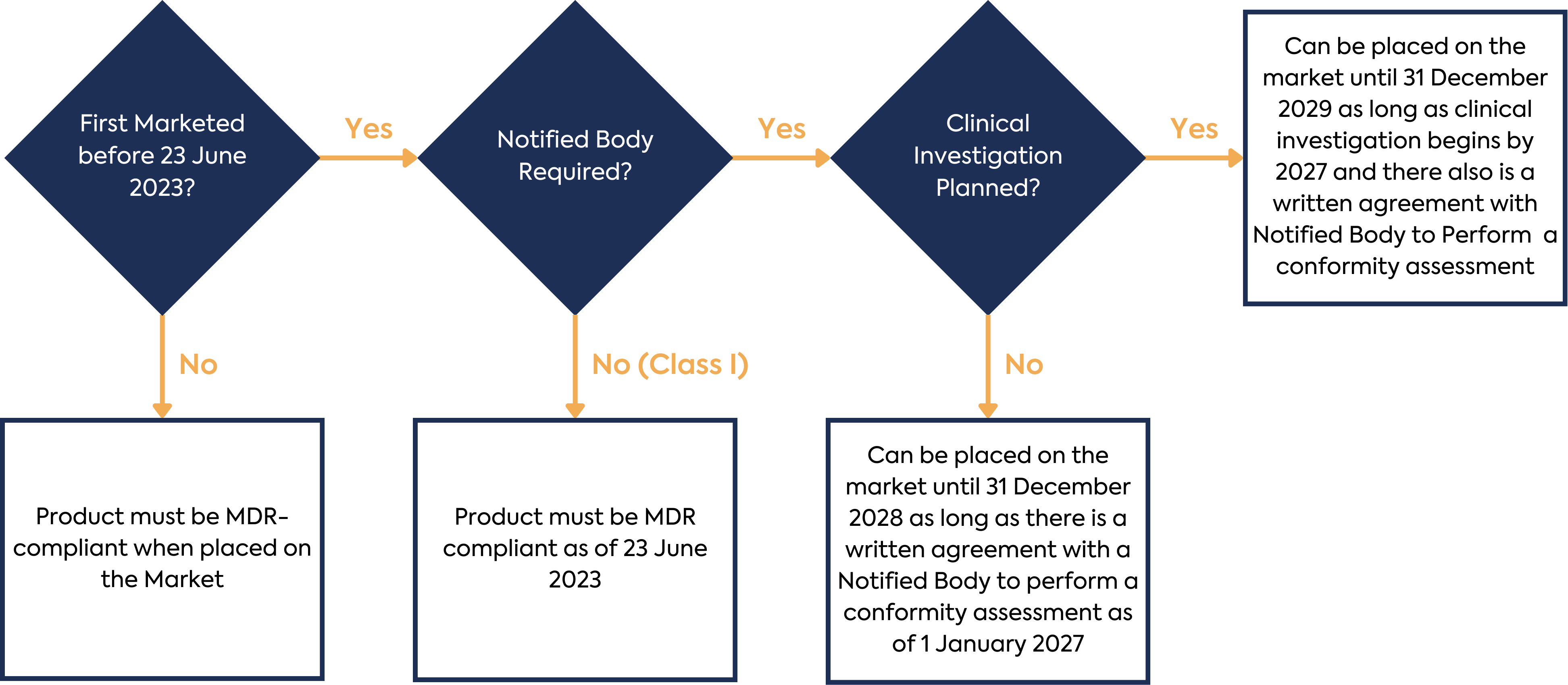
Once all the requirements of the MDR and applicable standards have been implemented, Annex XVI device manufacturers (with the exception of Class I device manufacturers) must engage with a Notified Body to gain certification for the device.
For manufacturers of Annex XVI devices, prior to the MDR, there were no specific requirements for regulatory compliance or certification in the EU. Annex XVI device manufacturers must now consider how to retroactively apply the MDR to existing products to ensure compliance or navigate the different requirements for developing new Annex XVI devices.
Implementation of the MDR is difficult without prior knowledge of the medical device legislation in the EU. Expertise is necessary on how to interpret and apply the rules within the MDR to the device and the organisation itself. Engagement with QMS, risk management, product, and regulatory professionals, such as DLRC, can assist with streamlining the process to ensure continued or new regulatory compliance for these device types.


Published May 12, 2026

Published May 07, 2026

Published Mar 20, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025