Using Out-of-Specification ATMP Batches: EMA’s 2026 Q&A
Published Mar 20, 2026
Published 01st July 2024

The EU (European Union) Health Technology Assessment (HTA) Regulation (EU Regulation 2021/2282) was adopted in 2021 to harmonise the clinical evaluation of health technologies across member states (MS) and improve efficiency by reducing the duplication of assessment across HTA bodies. Ultimately, the consolidation of a centralised assessment should improve access to medicines across Europe.
A single EU-wide assessment should, in theory, reduce the current duplication of work for both health technology developers (HTD) and HTA agencies that results when manufacturers are required to submit multiple clinical dossiers in multiple EU MS, ensure efficient resource use, and strengthen the quality of HTAs.
However, the complexities for HTD of compiling an EU-wide dossier submission during the period of regulatory review of the related marketing authorisation application (MAA) and submitting an HTA Joint Clinical Assessment (JCA) dossier at the latest 45 days prior to CHMP opinion need to be carefully considered. Efficient planning of resource requirements and key resource availability and sharing of information between global, regional and local regulatory and market access teams will be critical to ensure success.
Read on for an overview of the EU HTA Regulation, progress towards implementation, and key considerations for cross-functional collaboration to optimise HTA dossier submission and timely JCA conclusions.
The HTA Regulation is a formal continuation of the European Network for Health Technology Assessment (EUnetHTA) and requires the conduct of Joint Clinical Assessments (JCA), Joint Scientific Consultations (JSC), horizon scanning activities, and voluntary cooperation in areas outside the mandatory scope of the Regulation. These activities will be overseen by the HTA Coordination Group (HTACG) composed of representatives of HTA authorities and bodies from all EU MS.
The HTA Regulation will cover all medicines approved via the centralised route, within the mandatory scope, in-vitro diagnostics (IVD), and high-risk medical devices. A phased rollout of the Regulation for medicinal products means new active substances for oncology indications and advanced therapies will be subject to JCA under the Regulation from 12 January 2025. Progressive implementation will lead to the inclusion of Orphan products in 2028 and, finally, in 2030, all remaining medicinal products in the scope of the Regulation.
With respect to medical devices and IVD, the European Commission will adopt a decision on products in scope for JCA based on criteria such as unmet medical need, first in class, incorporation of software using artificial intelligence, machine learning technology or algorithms, and major Union-wide added value.
One of the key principles of the HTA Regulation is the inclusion of input from the HTA stakeholder network, including patient associations, non-governmental organisations in the field of health, and health professionals. The HTACG will facilitate dialogue between stakeholder organisations in assessing the dossier submitted by the HTD to support the JCA.
The Commission Implementing Regulation ((EU) 2024/1381) laying down the procedural rules for JCA of medicinal products was adopted in May 2024, as well as the template for the JCA dossier.
The JCA process begins at MAA filing, with the aim that this joint assessment of safety and clinical effectiveness will run in parallel to the MAA review by EMA, as shown in Figure 1. The deadline for submission of the JCA dossier is 45 days prior to CHMP opinion, and the final report on clinical effectiveness will be published 30 days following Marketing Authorisation (MA) – thereby expediting the final pricing and reimbursement decisions nationally.
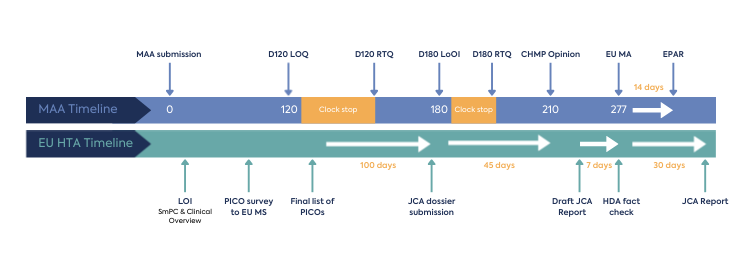
Considering the time needed for the scoping process and JCA dossier creation, operationalisation of the EU HTA process under the reduced timeframe for would be an additional challenge for HTD.
The JCA process is initiated by the letter of intent submitted by the HTD, which will include the Summary of Product Characteristics proposed by the applicant and the Clinical Overview section of the MAA (Module 2.5). This triggers the HTACG to send out a scoping survey to all EU MS covering information for the PICO (population, intervention, comparator, outcomes) framework.
Each MS will have 2 weeks to respond to the survey. It is not clear what the impact will be should any MS not be able to meet the 2-week timeline for the survey, I.e. whether they would be eliminated from the JCA process and the potential downstream impact of their non-inclusion.
However, the Regulation states that the scoping “must be inclusive of and reflect member state needs”, indicating a requirement to encompass all potential EU27 patient populations and treatments. This could raise considerable complexity and challenges in the process.
Based on the scoping survey results, the JCA assessor will consolidate the PICO survey results. The consolidation process aims to create a list of the lowest possible number of PICOs, which will form the basis of the JCA dossier to be compiled by the HTD. This can be particularly challenging with oncology, where there are multiple combination therapy recommendations and variable standards of care (SoC).
The HTD will have limited initial input into the scoping process. HTD can request a meeting with the JCA subgroup following receipt of the assessment scope. Any meeting between the HTD and JCA subgroup will take place no more than 20 days after the subgroup has finalised the assessment scope.
The consolidated list of PICOs forms the basis of the JCA dossier and will be shared by the HTACG. The developer will have approximately 100 days to arrange the submission of the JCA dossier (60 days for an MAA under accelerated assessment). The deadline for submission of the JCA dossier submission must not exceed 45 days prior to CHMP’s opinion on the MAA. This could be a demanding timeline for HTD to create a robust evidence package based on the consolidated PICO. To optimise preparation, HTD will need to consider forecasting the outcome of the consolidated PICO through collaboration with their local EU affiliates in order to understand their anticipated local PICOs and potential EU-wide consolidation.
The JCA will cover safety and clinical effectiveness only. It will not cover value judgements, economic analysis, or reimbursement decisions, which will continue to be evaluated by each MS HTA body. Therefore, although the JCA is mandatory, overall market access decision-making will remain at a national level.
There is limited opportunity for dialogue during the assessment of the dossier. JCA assessors can request missing information or evidence, and the HTD will only have 15 days to respond (10 days for MAA under the accelerated procedure and 14 days for medical devices or IVD). There is no clock stop in the JCA process. Additional requests for clarification may also be issued at any time during the preparation of the JCA report; in these cases, the HTD will have 7-30 days to respond, depending on the nature of the information requested. Any anticipated new clinical data should be indicated at the submission of the dossier, with the expected timeline for the new data availability.
If the therapeutic indication changes during the MAA’s assessment, the EMA should inform the JCA assessor and assess whether the change affects the assessment scope. Procedural rules for HTACG cooperation with the EMA are expected to be released in Q3 2024.
A decision will be made to determine whether the current JCA can continue or should be restarted. The HTACG will inform the HTD of this decision, and the PICO scoping exercise may be re-initiated. This eventuality would clearly have a considerable impact on the HTD, given the requirement and timeline for a revised JCA dossier. The impact on the timeline for the availability of the JCA report following the EC Decision on the MA is unclear at this stage.
Once a draft JCA report is provided, the HTD has 7 days to provide a factual check (5 days for products under accelerated assessment).
The JCA report will be published 30 days following the EC Decision on the MA.
MS are only required to give “due consideration” for the JCA report published and draw their own conclusions from the report. This allows MS to request further information from the HTD (including additional PICOs) to finalise their own evaluation and could lead to inconsistent approaches to market access evaluation. The JCA could lead to more restrictive access in some markets. For example, where there are subpopulations for a target medicine eliminated during the centralised PICO consolidation, but where the MS does not request additional information from the HTD. Thus, the influence of the JCA report on individual MS decision-making for pricing and reimbursement is unclear.
Where the JCA specifies the need for an update, the HTD should inform the HTACG when additional evidence for further assessment becomes available. Alternatively, HTDs may proactively share new relevant information, data, analyses, and other evidence with the HTACG. Based on the additional information provided, the HTACG will decide whether to include a JCA update in its annual work programme. As far as possible, the update to the JCA will be conducted by the same assessor, co-assessor, and external stakeholders involved in the initial assessment.
JSC will consist of scientific advice provided jointly by HTA bodies to HTD on their evidence generation plans, which can be provided in parallel with regulators. The aim of the JSC is to support the generation of optimal and robust evidence that satisfies the needs of HTA bodies and ultimately facilitates patient access.
Health technologies that are likely to be eligible for JCA are eligible for JSC. JSC will not be legally binding upon the HTD, HTACG or EU MS. However, the implications for HTD of not following JSC advice are not clear and should be carefully considered and justified, as would be the recommendation for regulatory-only scientific advice.
Where the number of requests for JSC exceeds the number that can be resourced, the HTACG will select applications according to unmet medical need, first in class, the potential impact on patients, public health, or healthcare systems, significant cross-border dimension, major Union-wide added value, or Union clinical research priorities (Figure 2).

Due to the limited capacity of JSC, not all innovative therapies will receive joint advice. For those HTDs not selected for JSC, the alternative may be to approach national HTA bodies for advice.
Implementing acts for JSC medicinal products are expected to be released for consultation in Q3 2024 and for medical devices by Q4 2024.
The implementation process for the HTA Regulation is still ongoing and companies will need to keep a close eye on the guidance expected to be published over the next 6 months according to the implementation rolling plan. Ongoing consultations provide insights into the anticipated process and engagement with local HTA bodies to iron out the challenges.
Preparing for the HTA Regulation means considering changes in company practice. HTD will need to map out their processes and define their governance model considering the cross-functional approach for the JCA dossier. Current templates will need to be reviewed to find a consolidated approach to input into the JCA dossier and align it to the required JCA dossier format.
As the JCA process will run in parallel to the MAA procedure, increased collaboration between regulatory and market access teams to ensure alignment in key messages between the MAA and the JCA dossiers will be essential. The complexity of providing the JCA dossier 45 days prior to CHMP opinion will have varying levels of impact on HTD and brand teams, depending on their resourcing, competing priorities and internal processes. Market access teams will need to flag products that will likely require complex HTA early in the development process to ensure sufficient time and resources are available for the generation of the dossier.
Consultation with local affiliate medical and market access functions for input on local PICO will be essential in order to anticipate the potential consolidated list of EU PICO. Creating an evidence package that meets the needs of all MS will be challenging, e.g. if there is significant local and historical variation in the treatment approach. This means that early engagement with local staff and their key external stakeholders (for example HCPs and patient groups) to understand the populations and comparators relevant to payers in each MS will be crucial for global evidence generation plans.
With the clinical effectiveness evaluation running in parallel to the MAA, alignment is needed on how potential changes in the wording of the indication will be handled and aligned in both MAA and JCA workstreams, their impact on PICO, and their impact on the overall HTA timeline.
The harmonised EU-wide JCA may also impact commercial launch planning for HTD. For example, MS, historically not considered to be “early launch markets”, may issue local HTA decisions within a shorter timeframe post-MA. The downstream effect of concurrent pricing and reimbursement decisions across MS may also put pressure and require additional planning in manufacturing and supply chain, affecting the traditional staggered market launch whilst manufacture is scaled up. The impact of earlier price differentials across MS could also lead to earlier parallel trade.
It will be critical for market access teams to analyse International Reference Pricing (IRP) networks and ensure target prices are achieved without unexpected impact from the pricing and reimbursement processes in other MS. It is possible that in certain circumstances, the challenges and complexities of the EU HTA Regulation and potential impact on IRP could actually deprioritise Europe in the global filing sequence.
Within the HTA Regulation, randomised controlled trials are the preferred comparator studies. Oncology and Advanced Therapy Medicinal Product (ATMP) studies can often be associated with difficulties in identifying the target comparator where there is variability in the SoC. Furthermore, with innovative therapies often targeting areas of particularly high unmet need, there is also an increase in the conduct of single-arm studies and the use of surrogate endpoints.
Embedding the JSC in the early development process and collaborating more closely between functional groups, including development, regulatory, and market access, will be necessary to optimise the incorporation of relevant PICO in the clinical study design and ensure robust HTA evidence generation.
To conclude, in preparation for implementing the HTA Regulation in 2025, procedures for conducting the JCA are being developed, and EUnetHTA 21 is authoring guidance and templates to facilitate the process. In tandem with JSCs, the JCA aims to speed up access to new therapies, reduce duplication of work, and harmonise methodologies used for clinical evaluation by different HTA bodies within the EU.
To achieve an optimal EU-wide HTA dossier submission, HTD will need to ensure engagement and cross-functional collaboration across organisational functions such as regulatory, market access, and development, foster robust networks, both internally and externally, with local and regional HTA stakeholders, and ultimately maximise the efficiency of both regulatory and HTA evidence generation.
DLRC works with HTA advisors to support companies in adapting to the new regulation, enabling cross-collaboration between market access and regulations. Get in touch to see how we can further support you in meeting the new regulations for 2025 and beyond. Email our team at hello@dlrcgroup.com or use the link below.


Published Mar 20, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025

Published Nov 25, 2025

Published Nov 24, 2025

Published Nov 24, 2025