The Evolving Landscape of EU Centralised Marketing Authorisations
Published May 12, 2026
Published 12th May 2026

The European Regulatory Framework for Medicinal Products continues to evolve in response to scientific innovation, unmet medical needs, and the complexity of modern pharmaceutical development. The recently agreed pharmaceutical legislation introduces significant changes to the centralised marketing authorisation (MA) process, the European Medicines Agency’s (EMA) role and organisation, and the obligations imposed on marketing authorisation holders (MAHs).
The centralised procedure allows the submission of a single marketing authorisation application (MAA) to the EMA. Approval under this procedure permits marketing throughout the EU and the European Economic Area (EEA). The reform of the European Union (EU) pharmaceutical legislation reshapes the centralised procedure to better align regulatory decision-making with public health priorities while maintaining a high standard of scientific evaluation. This article provides an overview of the changes introduced in the new EU pharmaceutical legislative overhaul, which will shape the future of medicines regulation in the EU.
The centralised procedure is increasingly positioned as the default access route for medicines of strategic value to the EU. The new EU reform aims to:
To support faster access and more efficient evaluations, the reform seeks to simplify and streamline the EMA’s structure while preserving depth of expertise and independence. Key objectives include:
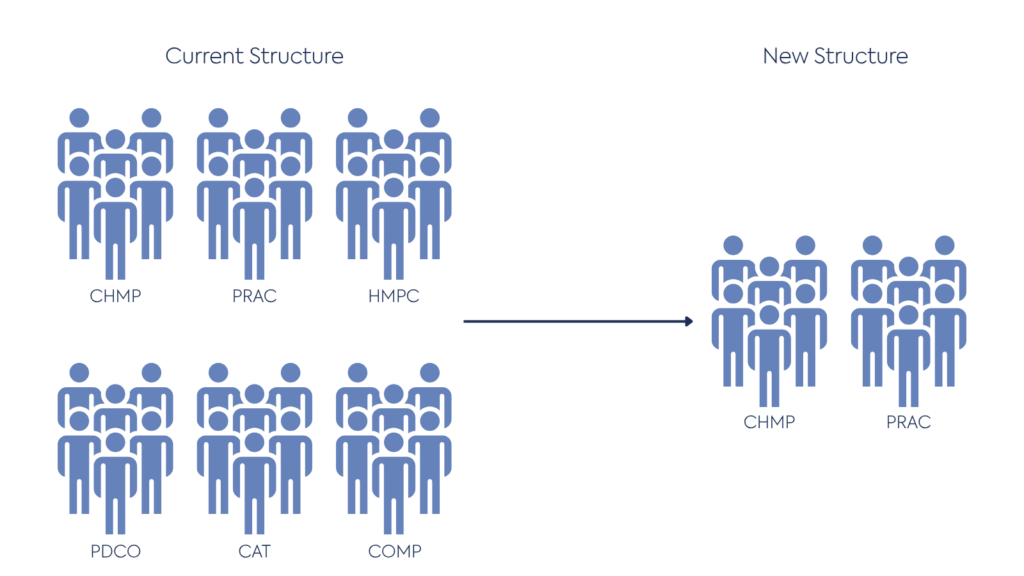
The current six committees will be streamlined to two main committees: CHMP and PRAC. The CHMP remains the central body responsible for evaluating quality, safety, and efficacy; the reformed framework supports greater coordination between working parties, clearer allocation of responsibilities and a new streamlined assessment structure.
The centralised procedure ensures harmonised scientific evaluation and consistent availability across all Member States. This procedure is mandatory for a range of medicinal products, especially those involving high technological medical products and areas of significant public health interest. In addition to products derived from biotechnological processes, advanced therapy medicinal products (ATMPs), orphan medicinal products, and products applying for paediatric-use marketing authorisations, the mandatory scope is expanded to include priority antimicrobials and any product containing a new active substance. These additions reflect new policy priorities around innovation, preparedness, and public health impact. The optional scope will remain available for other products such as generics, biosimilars and hybrid applications.
The EMA retains the ability to conduct phased (rolling) reviews of complete data packages for individual modules. This is applicable for medicinal products which are likely to address an unmet medical need and are expected to be of major interest from a public health perspective, certain antimicrobials, or products intended to be used in a potential or recognised public health emergency.
Where there is an unmet medical need, a conditional MA can be granted before the submission of comprehensive clinical data, provided that the benefit of the product’s immediate availability outweighs the risks. A conditional MA is valid for one year, renewable annually, and may be subject to specific obligations, such as completing or initiating studies to confirm a favourable benefit-risk.
Whilst under the old regulatory framework, conditional MAAs were only possible for a new MAA of a new active substance, under the new EU pharmaceutical legislation, a conditional MA may also be granted for new therapeutic indications of an already authorised medicinal product, where the same criteria as outlined above for a conditional MAA apply.
There are no changes to the regulations on exceptional circumstances. In such an event, an MA may be granted if comprehensive data cannot be provided to the Agency. An MA granted under exceptional circumstances must be reassessed after 2 years or as otherwise agreed.
Following the COVID-19 pandemic and gaps identified during this time, the Agency will introduce a new Temporary Emergency Marketing Authorisations (TEMA) specifically designated for Union-recognised public health emergencies.
This applies to products intended to treat or prevent the medical diagnosis of a serious or life-threatening disease or condition directly related to the public health emergency before complete quality, non-clinical, clinical, and environmental data are available. However, this does not apply to genetically modified organisms.
TEMA enables:
TEMA is explicitly time limited and transitional. Once sufficient data is generated, the MAH must submit a full MAA to replace the emergency authorisation. Crucially, regulatory protection periods are calculated from the TEMA grant date, not the later full MA.
MAAs submitted through the centralised procedure must be fully electronic and complete, which is anticipated to enable further use of AI. With the new EU legislation, there are changes to the dossier submission and particulars, which include:
The reform significantly shortens regulatory timelines:
The Agency retains a structured validation phase, during which it has 20 days from receipt of the application to confirm validity. During this period, the completeness of the dossier is assessed, and any missing information or critical deficiencies must be corrected within the Agency-defined timeframe. Applications with unresolved critical deficiencies may be terminated within 90 days of validation. The Agency will develop scientific guidelines for identifying “critical deficiencies” that may prevent the evaluation of a medicinal product.

Currently, MAs are granted for an initial period of 5 years from the date of first grant, after which a renewal is required. Under the new legislation, MAs will be granted for an unlimited duration, unless there are justified safety grounds, in which case one renewal may be required.
One of the most significant policy shifts is the strengthening of availability and supply obligations. While centrally authorised products have historically not been subject to EU‑level launch obligations, the new framework introduces:
This obligation must not jeopardise the company’s financial viability or be considered breached or exceptional circumstances (e.g., external supply disruptions beyond the MAH’s control). The MAH does not have to comply, provided they are justified, and the circumstances are documented.
Further details on shortage monitoring and security of supply will be discussed in another article.
Requirements around suspension, withdrawal and non-renewal are strengthened. MAHs must now:
The sunset clause remains in force. If a product is not placed on the market within three years of the MA grant, or if marketing ceases for three consecutive years, the MA may become invalid unless a justified exemption is granted.
Transparency obligations are expanded and reinforced, including:
All disclosures remain subject to the protection of commercially confidential information.
The revised EU framework for centralised marketing authorisation reflects a broader vision: ensuring faster access to safe and effective medicines, supporting innovation, and strengthening oversight throughout the product lifecycle. With clearer timelines, expanded scientific requirements, and new flexibility mechanisms, the regulatory environment is evolving to meet the demands of modern therapeutics and public health challenges.
DLRC offers dedicated expertise to support companies navigating the EU centralised marketing authorisation procedure, providing clarity and confidence at every stage. We can help understand whether the centralised route is mandatory or strategic for your product, interpret EMA and CHMP expectations, and anticipate the regulatory implications of evolving EU pharmaceutical legislation. This early strategic guidance ensures development and submission plans are aligned with centralised procedural requirements from the outset.
We can provide comprehensive support for the preparation of MAAs, coordinating the development and review of Modules 1–5 to ensure consistency, completeness and regulatory compliance. Our approach reduces the risk of validation issues and enhances the application’s overall robustness ahead of the CHMP assessment.
Throughout the procedure, DLRC delivers hands‑on submission and procedural support, acting as a central regulatory interface with the EMA. We manage submissions via EMA systems, track key milestones, support responses to CHMP questions and requests for supplementary information, and advise on post‑authorisation obligations and lifecycle planning. By maintaining clear oversight of the centralised procedure, DLRC enables clients to progress efficiently through assessment and into successful EU market access. Contact us today at hello@dlrcgroup.com to discuss how DLRC’s award-winning consultants can help you throughout your product’s approval process.

Published May 12, 2026

Published May 07, 2026

Published Mar 20, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025