Updates to the EU Pharmaceutical Reform 2026
Published May 07, 2026
Published 07th May 2026

After years of trilogue discussions and negotiations between the European Commission, European Parliament and European Council, the final EU Pharma Package texts have been published and are due to take force in 2028. The new Pharma Package comprises a new Directive (6367/26) and Regulation (6366/26).
The Regulation is a full replacement of key centralised authorisation/EMA and incentives regulation: it repeals Regulation (EC) No. 726/2004, Regulation (EC) No. 141/2000, and Regulation (EC) No. 1901/2006, and amends Regulation (EC) No. 1394/2007, and Regulation (EU) No. 536/2014. The Directive repeals Directive 2001/83/EC and Directive 2009/35/EC.
The key policy change areas include:
In this article and the series to follow, we will investigate the key changes to the current pharmaceutical framework and highlight the implications of these changes. It will be important to all stakeholders to prepare and adjust future strategy to account for new incentives and revisions to product protections.
The “8+2(+1)” exclusivity framework has been re-designed following changes to Articles 80-83 of the revised Directive. This leads to a maximum achievable market protection of 12 years (see a breakdown in the figure below for reference).
Regulatory data protection remains at 8 years following the granting of a marketing authorisation. In this period innovator data shall not be referred to by another applicant for a subsequent marketing authorisation. This period can be extended by 1 year if an exclusivity voucher is used in accordance with revised Regulation, Article 41(1) (transferable exclusivity voucher from the antimicrobial incentive).
Currently innovators receive a baseline of 2 years market protection, where a generic or biosimilar product cannot be placed on the European market, but regulatory preparation can take place for a launch during this period. Expanded Bolar exemptions will make this preparation easier for generic and biosimilars (more on this later). Under the revised directive this market protection is reduced to a 1-year baseline.
The revised Directive introduces incentives to extend market protection by +1 year in the following circumstances (Article 81 of the directive):
or
or
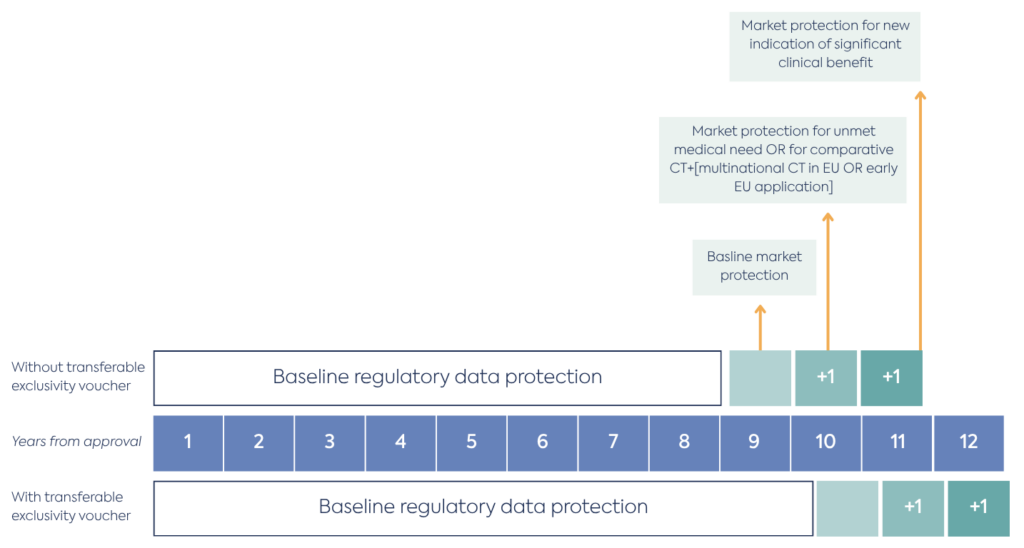
Therefore, the maximum protection period that a drug could have is the following: 8 years of data protection + maximum 3 years of market protection if the criteria laid above are met. An additional year of regulatory data protection can be granted if an exclusivity voucher from the new antimicrobial incentive is granted.
The revised Directive provides definitive text on what generics and biosimilar sponsors can prepare leading up to the expiry of innovator protections. Patent rights will not be infringed upon when these sponsor’s conduct the necessary studies, trials and other activities for the purposes of:
This offers transparency for generic and biosimilar manufacturers and enables their preparation following innovator protection expiry, for pan-EU launches earlier than before.
Medicinal product addresses an unmet medical need if at least 1 of its therapeutic indications relates to a life threatening or severely debilitating disease and either of the following conditions are met:
The revised Regulation introduces the concept of breakthrough orphan medicinal products mirroring the previous proposal for orphan medicinal products addressing ‘high unmet medical need’.
Criteria for breakthrough orphan medicinal products:
Applications based on bibliographic data will not be eligible for this breakthrough orphan medicinal product designation (Article 13 of the revised Directive).
The revised Regulation has changed the framework we know for orphan medicinal products significantly. Market exclusivity for orphan products offers protection against the authorisation of a similar medicinal product for the same therapeutic indication (unless specific derogations apply). Currently, orphan medicinal products receive 10 years of orphan market exclusivity +2 years upon completion of an agreed PIP. Additionally, where an applicant gets an extension of indication in another orphan indication, they receive a separate market exclusivity period of 10+2 years, for that specific indication.
Under the revised Regulation the duration of exclusivity shall be:
MAHs will no longer be able to hold separate market exclusivity periods for different MAAs of the same active substance. The duration of the market exclusivity shall start from the date when the first orphan marketing authorisation was granted in the Union and can be prolonged based on the criteria described below.
Orphan medicinal products (except those that were granted based on bibliographic data) can receive a prolongation of 1 year to their exclusivity if at least 2 years prior to expiration, the orphan MAH gets a MA for one or more new therapeutic indications for different orphan conditions. This may only be granted twice. Of note, prolongation of market exclusivity by indication extension is the only way to extend the protection period of orphan products.
The reform delivers regulatory tools to enable wider access to medicines to EU citizens. These include:
The publication of the final EU Pharma Package marks one of the most significant overhauls of the European pharmaceutical framework in decades. With the new Regulation and Directive due to be implemented in 2028, the landscape for data protection, market protection, orphan incentives, antimicrobial development, and generic/biosimilar entry will shift considerably.
Across the key policy areas examined in this article, several themes emerge:
The new framework will require all stakeholders, innovators, generic and biosimilar sponsors, investors, and healthcare systems to rethink development, protection, and access strategies. The shift toward performance‑based exclusivity means companies must plan earlier and more holistically for comparator selection, multi‑Member State trial execution, unmet‑need justification, and EU‑first filing.
For orphan products, development and lifecycle management strategies must be revisited to understand maximise exclusivity and assess whether additional indications remain commercially viable.
Meanwhile, the operational burden associated with supply obligations will demand robust regulatory governance and forward‑looking compliance systems.
These reforms introduce both opportunities and risks—and navigating them will require expert regulatory interpretation, strategic scenario‑planning, and detailed product‑specific assessment. DLRC is uniquely positioned to guide companies through this transition by offering:
To get in touch with our expects, email hello@dlrcgroup.com.

Published May 07, 2026

Published Mar 20, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025

Published Nov 25, 2025

Published Nov 24, 2025