EU HTA and JCA: Redefining Market Access Strategy in Europe
Published Feb 26, 2026
Published 29th November 2023
A Marketing Authorisation gives a pharmaceutical company permission to place a medicine on the market in a particular country or territory. Marketing Authorisations are granted by government appointed regulatory organisations, often referred to as health authorities or competent authorities. It is the role of health authorities to uphold strict safety standards for new medicines in accordance with legislative regulatory frameworks.
The European Union (EU) is a political and economic union consisting of 27 member states located in the European region. In the EU, many member states have their own nationally appointed health authorities, such as France’s National Agency for the Safety of Medicine and Health Products, and Sweden’s Medical Products Agency. In addition to EU member state national health authorities there is the European Medicines Agency (EMA). The EMA is a regulatory authority founded with the aim to harmonise pharmaceutical regulatory affairs in the EU and EEA (European Economic area) and achieve scientific excellence in the evaluation and supervision of medicines in the EU.
The EMA has jurisdiction across all 27 EU member states and EEA members, namely, Lichtenstein, Norway, and Iceland. The EMA harnesses the scientific expertise of EU national competent authorities for the benefit: risk assessment of medicinal products but it does not have the legal power to grant marketing authorisation in the EU (this is held by the European Commission). The European Commission is a politically independent group of commissioners who are responsible for proposing and enforcing EU legislation. The Commission was established in 1958 and it implemented the first law regulating pharmaceutical products in the EU 7 years later in 1965.
There are different types of legislation in the EU, some are legally binding, and some are not. Regulations and decisions are legally binding with regulations being applicable across the EU in its entirety whilst decisions are addressed to individual states or companies. A directive is another legislative act which is applied across the EU however, it is not legally binding but instead sets a ‘goal’ that must be achieved by all EU member states. The ‘goals’ set out in a directive will often give rise to the implementation of national laws that allow members to reach the goal and avoid financial penalties issued by the Commission.
European Council Directive 2001/83/EC, repealing Directive 65/65/EC, maintains that a medicinal product shall not be marketed without a valid marketing authorisation issued by a competent national authority. In the European Union, a medicinal product may be granted a marketing authorisation by one of 4 regulatory procedures, the national procedure, the centralised procedure, the mutual recognition procedure, or the decentralised procedure. These procedures have different scopes, applications processes, and outcomes, e.g., the number of EU member states that the medicine would be able to be marketed in.
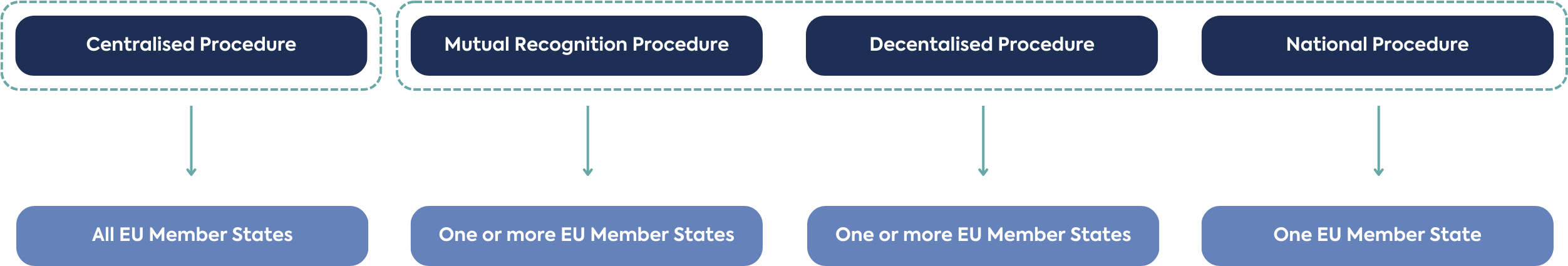
The national procedure predates the other marketing authorisation procedures. As originally laid out in Directive 65/65/EEC, national marketing authorisations are granted following an application to a single national competent authority, resulting in approval to market the medicine in that country alone. The directive details specific standards required of companies wishing to receive approval for their drug (see below list).
1. Company and Manufacturer Name and Address
2. Product Name
3. Qualitative and quantitative details for all constituents of the drug product
4. Method of Preparation
5. Indications, Contra-indications, and Side-effects
6. Dosage, pharmaceutical form, route of administration, expected shelf-life
7. Manufacturing controls and analytical methods
8. Results of pharmacological and toxicological tests
9. Results of clinical trials
10. Product packaging mock ups
11. Documentation of manufacturing site authorisation
12. Details of marketing authorisation applications in member states or third countries
Following the arrival of additional routes to marketing authorisation in the EU, the National Procedure is less widely utilised and often only in specific situations. With the introduction of the Centralised Procedure (discussed later) and the requirement that certain categories of medicinal products have to use the procedure, it is no longer possible to achieve marketing authorisation via the National Procedure for certain medicines. Although use of the National Procedure is waning, the regulations that govern the procedure are relevant to both the MRP and DCP since both these procedures involve the issue of national licenses.
Directive 2001/83/EC lays out the mutual recognition procedure, which offers the opportunity to obtain a product license in more than one EU member state based on an existing reference marketing authorisation. The decentralised procedure was first laid out in Directive 2004/27/EC and relies on the same recognition principal as the mutual recognition procedure. The decentralised procedure allows for a single marketing authorisation application to be made in multiple EU member states at once, where an EU national authorisation does not already exist. A decentralised application is evaluated by the elected reference member state, and, at the end of the assessment, the concerned member state(s) review a draft assessment report, Summary of Product Characteristics and labelling texts, before confirming whether they approve the application.
Post-authorisation variations are managed in the same way for both mutual recognition and decentralised procedure products. These submissions fall under the remit of the variations Regulation (EC) No 1234/2008.
Commission Regulation (EC) No 726/2004 sets out a pathway for obtaining an EU-wide marketing authorisation via the Centralised Procedure. The centralised procedure offers a harmonised regulatory pathway for marketing authorisation in the EU, with many benefits over the other procedures, including simpler post authorisation maintenance and simplified access and product branding across the entire EU market. The centralised procedure does not come without disadvantages, these include higher costs and a higher strategic risk, where rejection by the assessment committee would prevent the applicant from marketing their drug in all EU member states.
Certain types of medicines fall under the mandatory scope of the centralised procedure, e.g., new active substances for the treatment of Cancer or HIV/ AIDs and viral disorders. Some products can use the centralised, mutual recognition, or decentralised procedures, such as new active substances for the treatment of diseases outside the mandatory centralised procedure’s scope, or medicines of significant innovation and interest to EU-wide public health. Other medicines fall under the mandatory scope of the national, mutual recognition, or decentralised procedure’s scope, for example, some over the counter generic medicines use the national procedure.
The Committee for Medicinal Products for Human Use (CHMP) is an EMA committee that is responsible for the initial assessment of Centralised Procedure marketing authorisation applications, assessment of variations and extensions to existing marketing authorisations. They also consider recommendations from the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC). PRAC assesses the safety of marketed medicinal products for human use and, following their recommendations, the CHMP can advise that the European Commission make changes to the marketing authorisation.
Following a positive opinion from the CHMP regarding a request for marketing authorisation or another applicable submission, the EMA will make a recommendation to the European commission that they grant the requestee the marketing authorisation or other request. The European Commission’s agreement with this recommendation is called the ‘decision’ and it marks the effective date for the requested submission.
The CHMP will evaluate a medicine’s quality, compliance with international good practice standards, mechanism of action, pharmacokinetics, efficacy in target patient group, side effects, risk management plan, details of future follow up studies, and product information before recommending that the European Commission grants a marketing authorisation.
With the introduction of newer, more harmonised marketing authorisation application procedures there has come many benefits for applicants and patients due to simplified and harmonised approval pathways improving access to medicines across the EU region. These different authorisation routes also offer additional complications for applicants. Navigating the intricacies of the four pathways either pre-authorisation or post-authorisation without an experienced regulatory affairs team can impact the speed and success of a marketing authorisation application (and hence availability of new therapies in the European market) and can lead to compliance issues and potential disruption to patient supply.
DLRC has extensive experience of successfully supporting global Marketing Authorisation Applications from pre-submission interactions through to product approval. Whether you are a large pharmaceutical or a small biotech company planning to register your first product in the US, UK, EU or any other RoW region, DLRC has the capabilities to help bring your product to market. To find out how our experts can support you, email our team at hello@dlrcgroup.com or use the links below.


Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025

Published Nov 25, 2025

Published Nov 24, 2025

Published Nov 24, 2025

Published Nov 24, 2025

Published Nov 14, 2025