The Evolving Landscape of EU Centralised Marketing Authorisations
Published May 12, 2026
Published 12th April 2024
On 5th March, the Medicines and Healthcare products Regulatory Agency (MHRA) held a webinar on the MedTech Regulatory Reform in the UK. The webinar covered updates to the scope of the UK Medical Device Regulations (MDR), changes to the Classification of General Medical Devices, In Vitro Diagnostic Devices (IVDs) and Software as a Medical Device, and lastly changes to Essential Requirements.
Eve Hutchinson, Head of Regulation at the MHRA kicked off the webinar with an overview. Between September and November 2021, the MHRA conducted a public consultation on proposed changes to the regulatory framework for medical devices in the UK. The government response to this consultation was published in June 2022 and is available here. The MHRA have recently produced a roadmap outlining the next steps towards the future regulatory framework for medical devices in the UK. The general elections next year may impact the timings in the current roadmap.
The MHRA are now drafting the Statutory Instruments (SIs) for the regulation of medical devices in the UK, taking on board the comments from the consultation. The draft text will be published by the World Trade Organisation toward the end of 2024, following this it will be debated in parliament before it is intended to come into effect in 2025.
The overall objectives for the UK regulation are to improve patient safety, to align with international best practices, and to promote the UK as a favourable market for MedTech companies.
The future regulation will update the definition of general medical devices to include the following products:
The new definition will also use the term ‘disability’ rather than ‘handicap’.
The definition of ‘intended purpose’ will be amended. It will be set by the manufacturer from the standpoint of an objective observer, and it will be described in appropriate language for the intended users. This will be paired with new legislation on claims, to minimise misconceptions on the intended use of the device.
The definition of IVDs will include ‘software’. Software as a medical device (SaMD) is a set of instructions that processes input data and creates output data. There will be additional pre-market essential requirements for SaMD (refer to the consultation document for further information).
Products without an intended medical purpose, e.g. dermal fillers, and coloured contact lenses, have a similar patient risk profile to medical devices and will therefore be covered in the Future Enhancement Regulation.
The classification of general medical devices will change to reflect updates in technology, levels of risk, and to align with established international practice. Non-invasive devices that come into contact with mucous membranes and injured skin e.g. special dressings, will fall into Class I, IIa or IIb. Non-invasive devices used in the process of in vitro fertilisation (IVF) and assisted reproductive technologies (ART) will fall into Class III. The classification for invasive devices will be stricter, for example those delivering drugs by inhalation will fall into Class IIa or IIb, and the majority of implantable devices will fall into Class III (e.g. surgical mesh, joint replacements, and spinal implants).
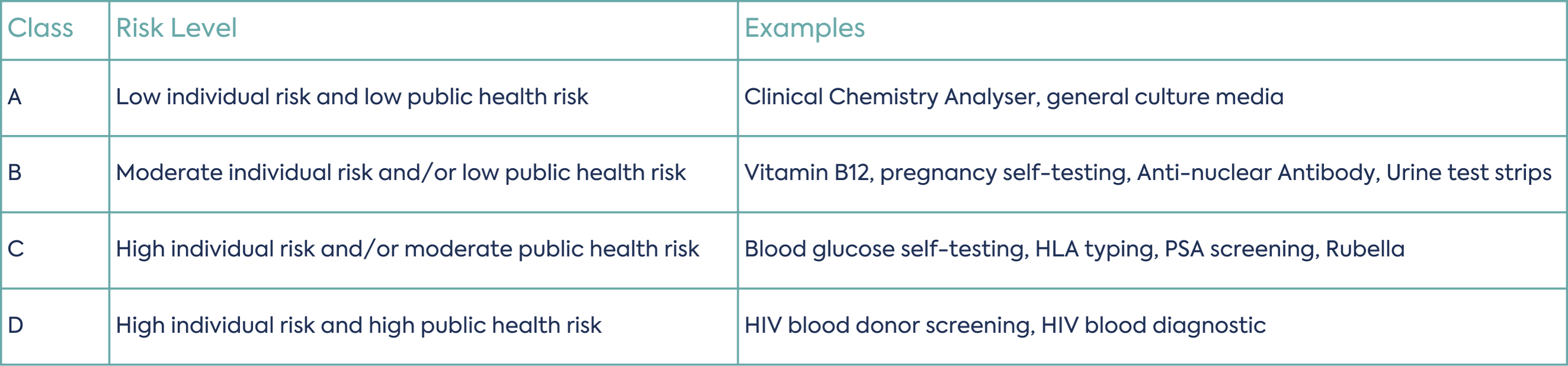
The classification of IVDs will align with the IMDRF approach (refer to N64 Principles of In Vitro Diagnostic (IVD) Medical Devices Classification). The classification will depend on the intended purpose, intended user, and the impact of the result to the individual and/or public health. There will be seven rules to determine the device class. A four-class system is proposed, with Class A being self-declared and Class B, C, and D requiring conformity assessment. Note that the new Classes are for IVDs only, therefore general medical devices will remain as Class I-III.
Essential requirements will be aligned with EU General Product Safety Requirements (GPSRs) as per the EU MDR and EU IVDR. There will be minor changes to make them specific for the UK, for example, referring to UK rather than EU law.
The information provided with devices generally includes labels, packaging, Instructions for Use (IFU) and electronic IFU (eIFU).
Unique Device Identifiers (UDIs) will be introduced for increased traceability, with additional specific requirements for Class III and implantable devices. Further details will be required on the clinical benefits of the device, shelf-life, storage conditions, and biosafety. Additionally, there will be increased measures to support environmental sustainability e.g. eIFUs for some devices; single copy IFU if multiple devices are supplied to the same user or location.
In summary, the future UK Medical Device Regulations will mostly align with Regulation EU 2017/745 (MDR) and Regulation EU 2017/746 (IVDR), facilitate access to innovative medical technologies and foster international harmonisation, allowing swifter access for devices approved by comparable regulators.


Published May 12, 2026

Published May 07, 2026

Published Mar 20, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025