Strategic Divestments: Maximising Product Lifecycle Value
Published Oct 21, 2024
Published 29th September 2023

Choosing novel trial endpoints for rare disease development has always been challenging for regulators and developers alike, but the global trend of increasing investment for new treatments for rare diseases and incredible advancement in gene and cell therapies mean that challenge is now amplified. As part of PDUFA VII, FDA have continued its commitment to advancing innovation by launching the Rare Disease Endpoint Advancement (RDEA) Pilot Program.
The RDEA Pilot Program will support novel efficacy endpoint development for drugs that treat rare diseases. The program will be jointly administered by FDA’s Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) during fiscal years (FYs) 2023 to 2027. Sponsors can begin to submit RDEA program applications starting 1 July 2023. A maximum of 13 proposals will be accepted by FDA over the duration of the program – so competition for participation in the pilot is expected to be high.
Read on for an overview of the goals, eligibility requirements, selection criteria, and procedural information for RDEA applications, as well as the resources available to guide rare disease endpoint development.
Rare diseases are estimated to impact 30 million people in the United States (US), and a similar number in the European Union (EU). Finding effective treatments for the 7000+ identified rare diseases is a huge task – but a critical one as rare diseases are often chronic, progressive, serious or life-threatening, and a large proportion begin in childhood.
Challenges associated with development of endpoints in rare diseases include limited information on disease natural history and/or heterogeneity in disease presentation, and small clinical sample sizes – requiring sensitive endpoints for which there is often no regulatory precedence. Combined with the increase in development activity in the rare disease space observed over the last decade and in both advanced therapy research and digital health technology, these challenges in endpoint definition and in regulatory experience and capacity create cross-discipline issues within rare disease drug development.
The RDEA Pilot Program has been designed to help overcome these hurdles and fulfils a commitment under Prescription Drug User Fee Act (PDUFA) VII for FDA to advance and facilitate rare disease drug development programs by providing a mechanism for sponsors to collaborate with FDA throughout the efficacy endpoint development process. The RDEA Pilot Program aims to provide advice on how a proposed novel endpoint can be used in a specific rare disease drug development program. Sponsors admitted to the program may participate in up to four focused advice meetings with interdisciplinary FDA experts in addition to the associated review division.
In addition, the program is intended to promote innovation and evolving science, and to develop FDA staff capacity. Learnings on novel endpoint development will be shared through FDA presentations, guidance documents, public workshops and a public facing website. FDA has committed to conduct up to 3 public workshops to discuss topics relevant to the development of endpoints for rare diseases. This aspect is discussed further below in relation to sponsor disclosure requirements.
In order to be eligible for the RDEA Pilot Program, the associated development program should address a rare disease, with an active investigational new drug application (IND) or pre-IND. Sponsors that do not have an active development program but have, or are initiating, a natural history study where the proposed endpoint is intended to be studied are also eligible to apply. Proposals may also be considered for a development program for a common disease that includes innovative or novel endpoint elements, including the specific endpoint and/or the methodology being developed – if there is sufficient justification that the proposal could be applicable to a rare disease.
In addition, the proposed endpoint must be a novel efficacy endpoint intended to establish substantial evidence of effectiveness for a rare disease treatment i.e.an endpoint that has never been used to support drug approval or has been substantially modified from previous use to support drug approval.
FDA will give selection preference to proposals that have the potential to impact drug development more broadly, such as use of a novel approach to develop an efficacy endpoint or an endpoint that could potentially be relevant to other diseases; to proposals that collectively reflect a range of different types of endpoints; and to proposals that have novel approaches for collecting additional clinical data in the premarket stage to advance validation of the endpoint for a surrogate endpoint proposal. (If the sponsor is proposing to develop a surrogate endpoint as part of a rare disease application, participation in a prior Type C Surrogate Endpoint meeting with FDA is encouraged).
The RDEA proposal must be succinct – no longer than 12 pages in length – and content and format requirements are available on the FDA website.
RDEA proposals may be submitted throughout each FY quarter for consideration in the subsequent quarter (submission deadlines 31 December, 31 March, 30 June, 30 September). FDA will accept a limited number of proposals for admission into the RDEA Pilot. For FY 2023, sponsors may submit RDEA proposals beginning in the fourth quarter (1 July 2023; deadline 30 September 2023), and FDA will accept a maximum of one proposal. For FYs 2024 to 2027, FDA will accept up to one RDEA proposal per quarter with a maximum of three proposals per year. A maximum of 13 proposals will be accepted by FDA over the duration of the Pilot Program.
Within 60 days after the quarterly submission deadline, FDA will review the proposals, select a primary proposal to proceed to disclosure discussions and an alternate (back-up) proposal, and notify all applicants of their proposal status.
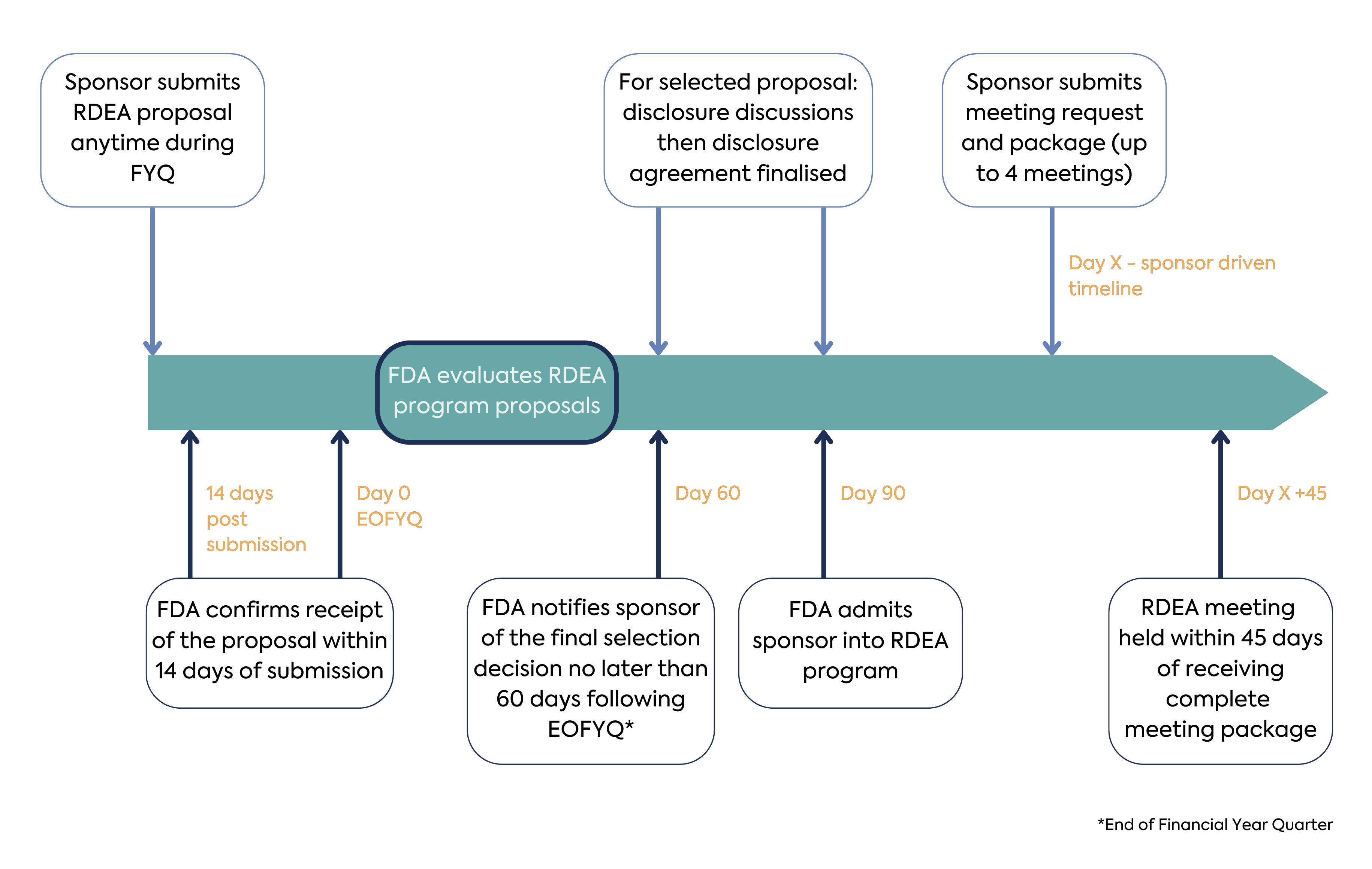
As described above, to support innovation and evolving science, novel endpoints developed through the RDEA Pilot Program may be presented by FDA as case studies, including while the drug studied in the trial has not yet been approved or biological products that have not yet been licensed for a given indication. FDA will aim to focus case study presentations on information that advances efficacy endpoint development and those elements relevant to understanding the novel efficacy endpoint and its potential use in a clinical trial intended to support regulatory approval. As such, prior to acceptance onto the RDEA Pilot Program, sponsors must agree on the information that FDA may disclose publicly and provide a rationale for that which is considered undisclosable. Once agreement is reached between FDA and the sponsor on the information that FDA may share publicly, FDA will notify the sponsor of admission into the program.
Once a sponsor is admitted into the RDEA pilot program, each RDEA meeting will be scheduled within 45 days following FDA’s receipt of the meeting request and complete meeting package. Similarly to the RDEA proposal, content and format requirements for the RDEA meeting request and package are provided on the FDA website.
After completion of four RDEA meetings, sponsors can request other formal meetings (Type B/C/ Type C Surrogate Endpoint/D meetings) to elicit further input on their novel endpoint as appropriate. Sponsors that are not selected to participate in the RDEA Pilot Program, or that do not wish to make the required disclosures, also have the opportunity to interact with the FDA through these traditional channels.
On 7/8 June 2023, Duke Margolis Centre for Health Policy Public Workshop, under cooperative agreement with the FDA, held a virtual public workshop entitled “Novel Endpoint for Rare Disease Drug Development”. In addition to introducing the RDEA Pilot Program, a variety of FDA and external speakers presented and discussed case studies for rare disease endpoint development using digital health technology, biomarker surrogate endpoints, clinical outcome assessment and multi-component endpoints.
All recorded presentations and materials associated with the workshop are available publicly via the Duke Margolis Center for Health Policy website, including a comprehensive resource guide of FDA guidance relevant to rare disease drug development in general, and more specifically for digital health technology, biomarkers, clinical outcome assessment and multicomponent endpoints.
As mentioned above, alternative pathways exist under which sponsors may discuss endpoint development with regulators. EMA also provides opportunities for sponsors to discuss rare disease drug development, via scientific advice or protocol assistance (for medicines with orphan designation).
Novel methodologies for medicine development may be submitted for EMA review, which if successful will result in a “qualification opinion” on the acceptability of the proposed method for a specific use, evaluation of which includes a public consultation phase. If EMA considers that the rationale and data to support the novel methodology are not complete, “qualification advice” may be issued consisting of recommendations for further method development.
FDA has established a Pilot Program to support the development of efficacy endpoints for rare disease treatments. The new Rare Disease Endpoint Advancement (RDEA) Pilot Program offers additional engagement opportunities with the FDA to sponsors of rare disease development programs that meet specific criteria, including an active IND or pre-IND for a rare disease, or a natural history study where the endpoint is intended to be investigated.
Key features of the RDEA pilot program include up to four focused meetings with interdisciplinary experts in addition to the associated FDA review division, and sponsor disclosure requirements that should be carefully considered by applicants prior to proposal submission.
All of this means that we are ideally suited to discuss the potential applicability and utility of the RDEA program versus other options for regulatory engagement, and to help you navigate regulatory requirements for the critical development of medicinal products for rare diseases. To speak to our experts about how they can help you achieve your regulatory goals, email us at hello@dlrcgroup.com or use the link below.


Published Oct 21, 2024

Published Oct 18, 2024

Published Oct 10, 2024

Published Oct 07, 2024

Published Sep 10, 2024

Published Aug 30, 2024
Published Aug 19, 2024

Published Aug 09, 2024

Published Aug 08, 2024