EU HTA and JCA: Redefining Market Access Strategy in Europe
Published Feb 26, 2026
Published 06th March 2024
There is a growing need for early access of medicines for all patients at all times. Expanded Access Programs (EAPs), also called Compassionate Use Programs (CUPs) have potential for allowing patient treatment using unlicenced medicines, separate from clinical trials and prior to marketing authorisation in the patient’s country, when no other treatment option is available. Regulatory agencies have mechanisms in place for all product types at various stages of product life cycle to support healthcare by using EAPs/CUPs.
To enhance product development and speed to market, throughout the full life cycle, companies are now including EAPs alongside their clinical development, product license, reimbursement, and marketing distribution plans to maximise assets. This approach has many benefits for patients, physicians, and pharmaceutical companies. However, developing a strategy to support EAPs to strengthen existing development through to distribution plans, whilst navigating complex country-specific requirements can be challenging. Key factors to consider are explained below and how DLRC can deliver Regulatory Strategies for your product and portfolio.
There are many names to describe EAPs, including compassionate use, early access, individual or named patient program, special access, unlicensed medicines, market access, etc. Expanded access is a potential pathway for a patient with a serious or immediately life-threatening disease or condition to gain access to an investigational medical product (drug, biologic, or medical device) for treatment outside of clinical trials and prior to commercially available products, when no comparable or satisfactory alternative therapy options are available.
EAPs (in the USA and other countries including EU and UK) supply unlicensed medicines for the treatment of one (individual or named patient) and/or more patients (groups), for a specific indication, where there is an unmet medical need, per country. Often, treatment is limited to serious and life-threatening conditions, but not always. As requirements and eligibility criteria for access vary, it is necessary to individually evaluate the regulatory strategy for each product, per indication, per country, considering the phase of development.
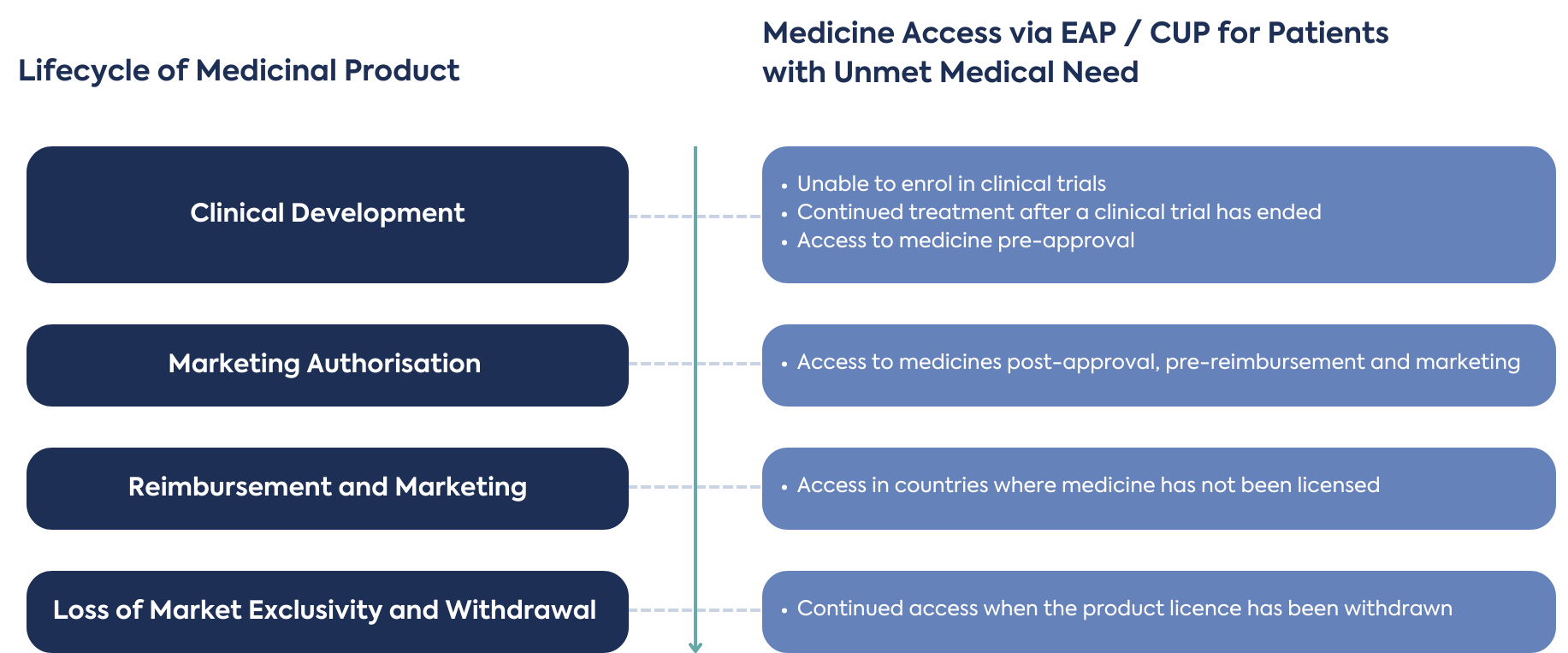
Although it is beneficial for EAPs to be planned and implemented early in the development process, there are opportunities throughout the full product life cycle, depending on eligibility criteria, sponsor and prescribing physician commitment of responsibility, benefits, and challenges to supply of unlicensed medicines. It may also be possible to provide new products in development to patients who did not meet the eligibility criteria to enrol in a clinical trial, or patients terminally ill with no other treatment options available.
EAPs have also been used for continued treatment of patients after a clinical trial has ended (post-clinical trial), and before the product is approved (pre-approval), reimbursed and marketed in a county. These processes are used to fill the supply gap, ensuring patients’ ongoing access to medicine. During this period, outside of a clinical trial, additional Real-World Data or Evidence (RWD/E) can be collected. New patients, not previously treated on clinical trials (naïve) may also receive treatment during a gap between product approval and launch, if eligible.
Many new product development strategies focus on a lead market for initial marketing authorisation, which is often the USA or EU, followed by a second phase of marketing authorisation applications in other countries. Once a product is licensed in the lead market, there are opportunities for EAPs to support patient access to medicines in countries where a marketing authorisation application for product licensing will not be submitted, is pending submission or approval, and where there is an unmet medical need.
Towards the end of a product life cycle, such as when a licensed medicine has been withdrawn, supply may continue to patients with no other treatment options. This can happen after the loss of market exclusivity when a product license has been withdrawn and creates opportunities to distribute for unmet medical need.
Therefore, it is possible for regulatory agencies to offer more than one mechanism for supply of unlicensed medicines per country, depending on the life cycle of a product, and eligibility criteria.
Firstly, the patients benefit from treatment they would not otherwise receive, particularly for serious, life-threatening conditions. EAPs may also support patient advocacy groups, creating a community and additional assistance for the patient and their families.
Physicians have the opportunity to treat patients with unapproved medicines, when no other treatment is suitable or available, supporting survival and quality of life. Physicians gain understanding and experience using the medicines and may continue to prescribe the treatment once it is approved. The physician may become a Key Opinion Leader (KOL), a trusted, well-respected influencer with proven experience and expertise in the therapeutic area. KOLs are able to assist pharmaceutical companies with their product development and provide opinions to regulatory agencies.
Pharmaceutical companies that plan EAPs early during product development will enhance their overall strategy, creating a cross-functional action plan that continues to achieve target objectives. Participating in EAPs allow early exposure of the drug with both patients and physicians.
Although EAPs often provide medicines free of charge, limited programs may allow reimbursement of the unlicensed supply.
A key advantage is the collection of data outside of clinical trials to gather Real World Data or Evidence (RWD/E). Depending on quality, the data may be used to support marketing authorisations and/or reimbursement, further product development and other post-approval improvements and promotions.
As there are different requirements per country, it is important to develop a clear multi-stakeholder regulatory strategy. This involves cross-functional alignment including, but not limited to regulatory, clinical, pharmacovigilance, manufacturing and supply, marketing, and the prescribing physicians. Before an EAP can begin, all eligibility requirements must be met, and regulatory approval granted by national regulatory agencies for unlicensed supply.
The pharmaceutical company (as Sponsor) and physician commitment responsibilities are key, especially to meet demand for supply for patients to receive access, costs, and regulatory compliance including safety reporting. The sponsor must ensure the unlicensed medicines are not promoted, seek opportunities for collection of RWD/E, and manage transparency of the programs.
Before an unlicenced medicine can be initiated via EAPs, the legal basis, eligibility criteria, and compliance to supply need to be planned and aligned with the company’s business strategy and timelines. A regulatory strategy will help navigate the complexities and challenges of the different and changing regulations in different countries. A detailed plan is needed to ensure a smooth process, avoiding potential risks and hurdles, and also help a company understand the impact of non-compliance on its operations and reputation. A product and country-specific plan can be created to analyse feasibility of meeting patient need, data collection, possibility of reimbursement, and steps from start to end of unlicensed supply. To ensure compliance, with adherence to all applicable regulations, the strategy will also need to be flexible to adapt to any changing circumstances and opportunities.
From product development to post-licensing, it is never too late to consider expanded access, as there are many opportunities throughout the entire product life cycle.
Your company may be developing and marketing medicinal products for orphan, rare, serious, or life-threatening disease or conditions where there are unmet medical needs. With benefits to patients, physicians, and pharmaceutical companies, reviewing the feasibility and creating regulatory strategy for an individual product or product portfolio will help patients by making medicines available.
A forward-thinking regulatory consultancy, DLRC understands individual company’s unique requirements for EAPs can provide the regulatory strategy find solutions for unmet medical needs. To discuss how EAPs can benefit and support your product development plans, contact our experts via the link below.


Published Feb 26, 2026

Published Jan 07, 2026

Published Dec 02, 2025

Published Nov 27, 2025

Published Nov 25, 2025

Published Nov 24, 2025

Published Nov 24, 2025

Published Nov 24, 2025

Published Nov 14, 2025