Innovating Non-Clinical Testing: NAMs and ESG Impact
Published Jul 21, 2025
Published 27th March 2025

The US regulatory landscape can be very difficult to navigate when developing and bringing a new drug, biologic or medical device to market. Nevertheless, sponsors can strategically engage with the FDA during product development through an Agency meeting. These meetings serve as valuable opportunities to receive guidance, clarify requirements, and discuss potential issues with FDA regulators.
There are several types of FDA meetings, tailored to drugs, biologics and medical devices. You can use these to ask for advice or address any previous feedback. To get the most out of your FDA interaction, it is essential that you craft your questions effectively in your request for a meeting. Carefully select your attendees when you schedule a meeting, and ensure everyone can participate in discussions and rehearse beforehand. Additionally, ensure you submit a briefing book to the FDA within the specified timeframe. The briefing book should be comprehensive, with sufficient background and data on the product and development program
Both drugs and biologics must undergo testing and approval processes to ensure their safety, efficacy, and quality. The regulatory frameworks for each are distinct:
A Type A meeting is for urgent issues to help a stalled product development programme’s proceed by discussing a new path forward. Examples include:
For example, sponsors may use a Type A meeting to discuss how their program has stalled while working to remove a clinical hold. FDA can review plans for a new direction in development and weigh in on the updated plans.
A Type B meeting usually occurs around major milestones, and Sponsors typically have one entitled meeting per milestone. These include:
This meeting is an opportunity to discuss development plans with the FDA, typically during the early stages of drug development. Getting early feedback from regulators is highly advantageous to the overall development strategy for a product.
A Pre-NDA/BLA meeting with the FDA helps to ensure that a submission for marking approval is in alignment with Agency expectations. This meeting can ensure that sponsors have comprehensive information to ensure there are no delays in approval.
EOP1 meeting topics include safety, tolerability, and PK/PD findings from the phase 1 study, and the protocol for phase 2. These meetings help to ensure the clinical data package and phase 2 study design will be acceptable to FDA.
The EOP2 meeting can be used to discuss the results from phase 2 and finalise phase 3 trial design. If phase 2 results are inconclusive, the FDA may advise on additional trials before proceeding. FDA feedback at this stage can prevent problems at phase 3 and registration.
This is any meeting other than a Type A, B, B (EOP), D, or INTERACT regarding the development and review of a product. Examples include:
An Advisory Committee meeting involves an independent group of experts who provide advice to the FDA on specific complex regulatory issues related to a product. However, the FDA does not make final decisions during these meetings. Nevertheless, they serve as an essential advisory step.
The Request for Designation meeting is used to clarify the appropriate regulatory pathway for a product (e.g., whether it should be classified as a drug, biologic, or device).
This meeting allows Sponsors to gain insight into whether a BTD request could be appropriate for their development program, and the optimal timing, prior to submission.
You can request meetings for Office of Orphan Products Development Designation programmes, Orphan-Drug Designation, HUD and Rare Pediatric Disease Designations. These meetings allow discussion of complicated designation questions that require extensive analysis with the Office of Orphan Products Development
A type D meeting has a narrow focus and is used when there is only one or two topics to address. You can request this at any stage of development.
INTERACT meetings are early in development and discuss novel and challenging issues prior to an IND submission, that may delay initiation or progress of IND enabling studies.

FDA guidance specifies the intended timeline for each meeting type, as summarised below:
| Meeting Type | FDA Response to Request | FDA Scheduled Meeting Date |
|---|---|---|
| A | 14 days | Within 30 days |
| B | 21 days | Within 60 days |
| B (EOP) | 14 days | Within 70 days |
| C | 21 days | Within 75 days |
| D | 14 days | Within 50 days |
| INTERACT | 21 days | Within 75 days |
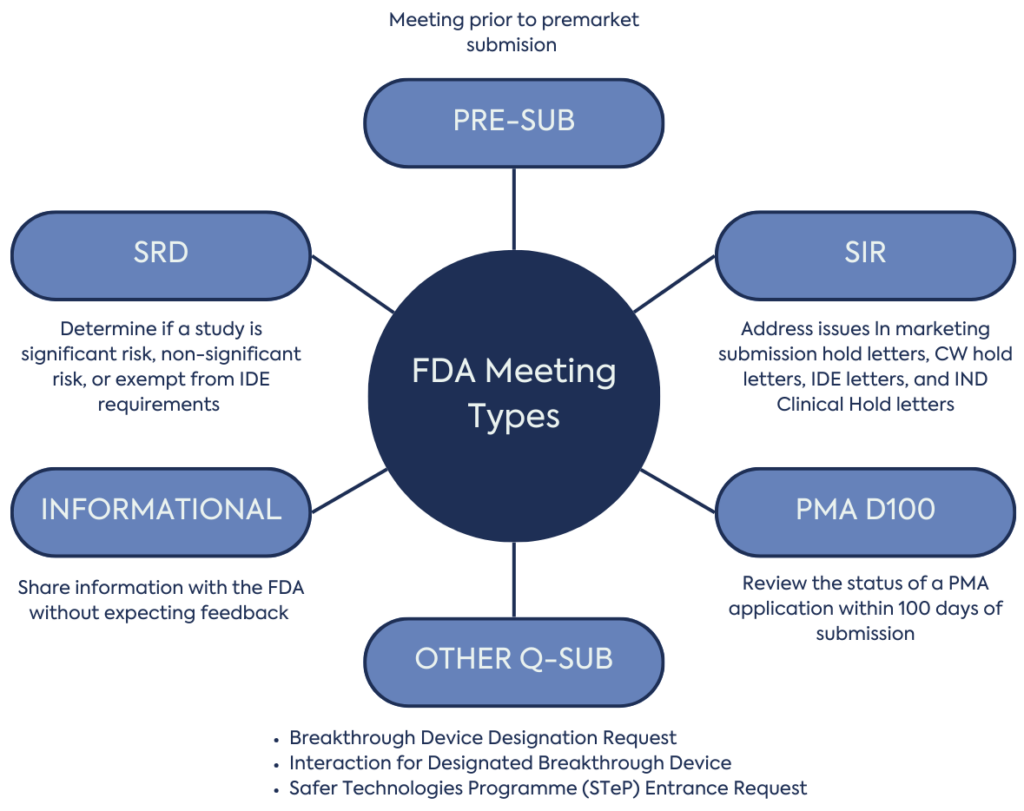
For Medical devices, FDA meetings are primarily organised through the Q-Submission programme by the Center for Devices and Radiological Health (CDRH) and CBER
This meeting allows manufacturers to meet with the FDA before a premarket submission e.g. Investigational Device Exemption (IDE), Premarket approval (PMA), De Novo request, 510(k). FDA expects the manufacturer to ask specific and focused questions, where Agency feedback would help guide product development and submission preparation.
These meetings help address issues conveyed in various letters, including marketing submission hold letters, CLIA Waiver (CW) hold letters, IDE letters, and IND Clinical Hold letters. The aim is to quickly resolve and clarify these issues, enabling submitters to address outstanding questions in their formal responses.
These meetings are used to determine if a clinical investigation for a medical device is significant risk, non-significant risk, or exempt from IDE requirements, helping to determine the level of regulatory oversight for the study and to facilitate IRB review.
These meetings are used to share information with the FDA without the anticipation of feedback. They are useful for familiarising the FDA review team with the device and providing an overview of its ongoing development.
These meetings review the status of a PMA application within 100 days of submission. They are a forum to discuss issues, remedial actions, and timelines. For major deficiencies, the PMA Day 100 meeting would be appropriate for asking clarifying questions or discussing the preliminary approach for resolution. Thereafter, submit a SIR to discuss the details of how to address a major deficiency letter
The Q-Submission Program also covers interactions from other FDA programs, such as the Breakthrough Devices Program (e.g. Breakthrough Device Designation Request; Interaction for Designated Breakthrough Device), and the Safer Technologies Program (STeP; e.g. STeP Entrance Request).
Additionally, some interactions that do not formally meet the definitions of the Q-sub types can be tracked using the Informational Meeting Q-Sub type. Feedback requests from the FDA that fall outside the scope of the Q-sub program can be addressed in other formal submissions or sometimes in informal interactions. DLRC can lead a productive engagement with the Agency and advise on the most suitable type of interaction.
| Meeting Type | Anticipated Meeting/Feedback Date |
|---|---|
| PRE-SUB | 70-75 days |
| SIR | 21 or 70 days* |
| SRD | 90 days |
| INFORMATIONAL | 90 days |
| PMA D100 | 100 days |
*Depending on whether SIR is submitted within 60 days or more than 60 days after issues were conveyed by FDA.
There are several steps you can take to get the most out of your meeting with the FDA. Firstly, it is essential that you identify your questions and an appropriate meeting type. Then you need to selectively decide your attendees and prepare a comprehensive meeting package to submit to the FDA. Prepare thoroughly, bring scientists and experts, and understand FDA language. Other tips include:
A client developing a novel, clinical-stage small molecule approached DLRC, wishing to expand their program to the US. The client wanted to arrange a meeting with the FDA to establish a good relationship with the Agency and de-risk their planned IND submission. Following a review of the client’s data package and objectives, DLRC advised that a pre-IND meeting would be appropriate for the immediate future (with scope for follow-up type C or D meetings on specific topics, if needed). Furthermore, DLRC assisted and supported all strategic and operational aspects of the interaction.
DLRC reviewed the client’s proposed list of questions and identified the specific Agency feedback and agreement needed at this stage to advance the client’s program, while also noting where clear guidance and precedent already existed. In turn, DLRC suggested a revised set of questions that would be more likely to culminate in a productive interaction with FDA.
For example, the client initially proposed an open-ended question on the tentative plan for a future registrational trial; however, DLRC suggested that a more phase-appropriate and focused question would be seek Agency feedback on the choice of endpoints. FDA’s response gave the client insight into the endpoints that the Agency would consider clinically meaningful. Thus this helped to de-risk the overall clinical strategy for the program. The client successfully interacted with the FDA, positioning themselves well to begin work on the IND submission and further advance the development of their innovative molecule for patients with high unmet need.
Navigating the regulatory landscape can be challenging, but FDA meetings can provide you with opportunities for feedback, guidance and clarity. DLRC can help you with strategic advice and guidance at all stages of development and help you approach the FDA. Knowing the type of meeting you need to progress your clinical development can seem daunting. Nevertheless, our extensive expertise can help guide you through this process. Find out how by contacting our regulatory experts at hello@dlrcgroup.com, or via the links below.


Published Jul 21, 2025

Published Jul 11, 2025

Published Jul 07, 2025

Published May 29, 2025

Published May 29, 2025

Published May 29, 2025

Published May 29, 2025

Published May 01, 2025

Published Apr 28, 2025